






Hosted By Metricstream

AI-Powered GRC Solutions Enabling Agility & Resilience

Accelerate Growth, Drive Risk-Aware Decisions
Reduce losses and risk events with forward-looking risk visibility. Enable a modern and integrated risk management approach with real-time aggregated risk intelligence and their impact on business objectives and investments.

Strengthen Compliance Posture, Prevent Risk of Compliance Violations
Protect brand reputation, lower the cost of compliance, and build regulators and board’s trust. Stay on top of evolving regulatory requirements, proactively manage compliance risks, policies, cases, and controls assessments.

Provide Better Assurance, Enable Agile Risk-Based Audits
Drive risk-aware decisions and accelerate business performance by aligning audits to strategic imperatives, business objectives, and risks. Provide timely insights on risks and strengthen collaboration across various functions.

Proactively Mitigate IT & Cyber Risks, Achieve Real-Time Visibility
Gain a quantified, unified view of risks, threats, and vulnerabilities in real-time. Proactively mitigate IT & cyber risks and ensure compliance with effective risk and IT control assessments and mitigation strategies.

Enable Growth with Purpose
Centrally manage requirements of various ESG frameworks including GRI, SASB, TCFD, and others, and optimize the process with automated data collection and reporting. Perform internal and supplier assessments as well as manage and mitigate risks.
AI Powered Solutions Enabling Agility & Resilience
Accelerate Growth, Drive Risk-Aware Decisions..
Reduce losses and risk events with forward-looking risk visibility. Enable a modern and integrated risk management approach with real-time aggregated risk intelligence and their impact on business objectives and investments.





Recognized as a GRC Leader by Industry Analysts

#12 in Chartis RiskTech 100 Report 2024
MetricStream Ranks #12 in Chartis RiskTech 100 Report 2024 and recognized as Category Leader in Enterprise Governance, Risk, and Compliance
Read moreLeader in Governance, Risk, and Compliance Platforms
MetricStream named a Leader in The Forrester Wave™: Governance, Risk, and Compliance Platforms, Q4 2023
Read more
Leader in Chartis Research GRC Solutions
MetricStream named a Leader in Chartis Research GRC Solutions, 2023 Market Update and Vendor Landscape
Read moreRecognized as a GRC Leader by Industry Analysts
MetricStream Ranks #12 in Chartis RiskTech 100 Report 2024 and recognized as Category Leader in Enterprise Governance, Risk, and Compliance
Read moreMetricStream named a Leader in The Forrester Wave™: Governance, Risk, and Compliance Platforms, Q4 2023
Read more
MetricStream named a Leader in Chartis Research GRC Solutions, 2023 Market Update and Vendor Landscape
Read moreHear From Our Customers

GRC Journey Award Winner

GRC Journey Award Winner

GRC Journey Award Winner

GRC Journey Award Winner

GRC Journey Award Winner

GRC Journey Award Winner

GRC Journey Award Winner
GRC Journey Award Winner

GRC Journey Award Winner

GRC Journey Program Excellence Award Winner






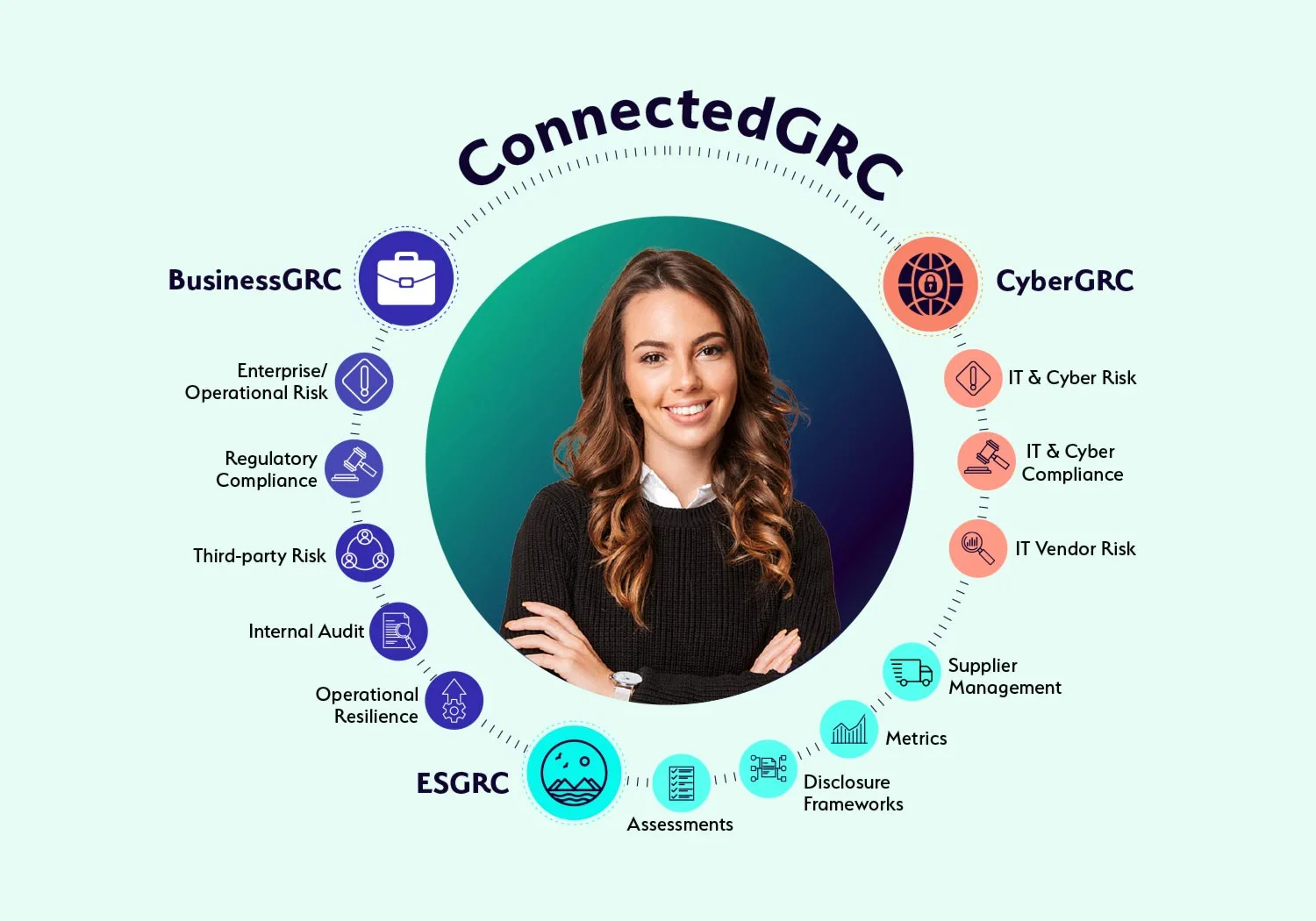
Why Choose a Connected GRC Platform?


Today’s dynamic enterprises require a connected to governance, risk, and compliance to better assess, manage, and mitigate risk across the enterprise. With MetricStream ConnectedGRC, your organization can:
Enhance decision-making with real-time insights into risk and compliance posture
Reduce risk exposure and losses, avoid risk of compliance violations, improve investments in growth, and gain a competitive advantage
Improve risk preparedness with better risk visibility and foresight through a proactive GRC approach






